Введен в действие
Постановлением
Госстандарта СССР
от 13 июля 1984 г.
N 2491
ГОСУДАРСТВЕННЫЙ СТАНДАРТ СОЮЗА ССР
КРЕМНИЙ ПОЛУПРОВОДНИКОВЫЙ
МЕТОД ОПРЕДЕЛЕНИЯ КИСЛОРОДА, УГЛЕРОДА И АЗОТА
Semiconductor
silicon.
Method of oxygen, carbon and
nitrogen determination
ГОСТ 26239.7-84
Группа В59
ОКСТУ 1709
Срок введения
с 1 января 1991
года
до 1 января 1991
года
Настоящий стандарт
устанавливает метод определения кислорода, углерода и азота в полупроводниковом
кремнии с использованием активации ускоренными ионами ![]() и протонами в интервалах значений массовых
долей примесей:
и протонами в интервалах значений массовых
долей примесей:
-7 -3
кислород инструментальный анализ от 5 х 10 до 1 х 10 %
-6 -3
с радиохимическим выделением от 1 х 10 до 1 х 10 %
-6 -3
углерод инструментальный анализ от 2 х 10 до 1 х 10 %
-6 -3
с радиохимическим выделением от 1 х 10 до 1 х 10 %
-5 -3
азот с радиохимическим выделением от 1 х 10 до 1 х 10 %.
Метод основан на
облучении анализируемых проб и образцов сравнения ускоренными ионами ![]() (определение кислорода и углерода) или
протонами (определение азота) с последующим измерением наведенной активности
радиоактивных изотопов
(определение кислорода и углерода) или
протонами (определение азота) с последующим измерением наведенной активности
радиоактивных изотопов ![]() и
и ![]() на спектрометре
на спектрометре ![]() -совпадений.
-совпадений.
Содержание примесей
в анализируемой пробе определяют путем сопоставления интенсивностей
счета импульсов радиоактивных изотопов определяемых элементов в пробах и
образцах сравнения.
1. ОБЩИЕ
ТРЕБОВАНИЯ
1.1. Общие
требования к методу анализа - по ГОСТ 26239.0-84.
2.
АППАРАТУРА, МАТЕРИАЛЫ И РЕАКТИВЫ
Ускоритель
заряженных частиц - источников ионов ![]() с энергией 13 мэВ при токе ионов в пучке 1 - 5
мкА, источник протонов с энергией не менее 6,5 МэВ и током ионов в пучке 1 - 5
мкА.
с энергией 13 мэВ при токе ионов в пучке 1 - 5
мкА, источник протонов с энергией не менее 6,5 МэВ и током ионов в пучке 1 - 5
мкА.
Спектрометр ![]() -совпадений
с кристаллами NaI(T1), имеющими размер не менее 100 х
70 мм.
-совпадений
с кристаллами NaI(T1), имеющими размер не менее 100 х
70 мм.
Универсальный
радиометр-дозиметр типа МКС-61Р.
Бюксы типа 1К-НЖ
для радиохимических работ с дополнительной защитой из свинцовых кирпичей и просвинцованного стекла в соответствии с требованиями
НРБ-76/87.
Средства
индивидуальной защиты от излучения и загрязнений радиоактивными веществами,
согласно требованиям ОСП-72/87.
Секундомер по ГОСТ
5072-79.
Весы аналитические.
Центрифуга
лабораторная со скоростью вращения 5000 об/мин.
Плитка
электрическая.
Штангенциркуль типа
В20034.
Насос водоструйный
лабораторный стеклянный.
Шайбы медные
диаметром 35 мм, высотой 13 мм с комплектом съемных алюминиевых диафрагм
толщиной 0,4 - 0,5 мм. Внешний диаметр диафрагм 30 мм, диаметры отверстий 7, 8,
9, 10, 11, 12, 13, 14 мм, по 5 - 8 шт. каждого размера.
Кассеты алюминиевые
диаметром 28 мм, высотой 5 мм. Толщина дна и крышки кассеты по 1,5 мм.
Фольга алюминиевая
толщиной 10, 15, 23, 34, 50, 70 мкм.
Асбест листовой.
Набор пробирок из
фторопласта-4 вместимостью по 20 см3.
Щипцы тигельные.
Тигли никелевые
диаметром 30 мм, высотой 50 мм.
Печь тигельная
вертикальная, диаметр кварцевой вставки 60 мм, высота 200 мм, мощность 1,5 кВт
или аналогичная печь.
Шланг резиновый
диаметром 6 мм.
Шланг
полихлорвиниловый диаметром 6 мм.
Силикагель для
хроматографии марки КСК с размером зерна 100 мкм.
Бумага
фильтровальная.
Бумага
полулогарифмическая по ГОСТ 334-73.
Калька.
Образцы сравнения:
пластины кварцевого оптического стекла марки KB размером 15 х 15 х 3 мм;
пластины, вырезанные из пирографита марки МПГ-8,
размером 15 х 15 х 3 мм; пластины нитрида алюминия размером 15 х 15 х 3 мм.
Пинцеты
металлические.
Пинцеты из
фторопласта-4.
Шпатели
металлические.
Абразивные порошки
М28, М20, М14, М10 по ГОСТ 3647-80.
Галлий
металлический технический.
Индий металлический
технический по ГОСТ 10297-75.
Чашки из
фторопласта-4, диаметром 30 - 35 мм, высотой 25 - 30 мм.
Реакционная колба
из молибденового стекла вместимостью 100 см3 круглодонная.
Пипетки из
оргстекла на 5, 10 см3.
Склянки Дрекселя вместимостью 50, 100 см3.
Стаканы стеклянные
вместимостью 50, 100, 500 и 1000 см3.
Фильтры Шотта N 4, диаметром 40 мм.
Колбы Бунзена
вместимостью 500 см3.
Цилиндры стеклянные
мерные вместимостью 25, 50 см3.
Цилиндры мерные из
оргстекла вместимостью 10, 15 см3.
Колбы стеклянные
мерные вместимостью 500 см3.
Пробки резиновые.
Кислота азотная по
ГОСТ 4461-77, концентрированная.
Кислота серная по
ГОСТ 4204-77, концентрированная.
Кислота
фтористоводородная по ГОСТ 10484-78, концентрированная.
Лантан
азотнокислый.
Натрий фтористый
технический.
Барий хлористый
технический по ГОСТ 742-78.
Аммиак водный по
ГОСТ 3760-79, х.ч., концентрированный и 25%-ный.
Натрий азотнокислый
технический по ГОСТ 828-77.
Натрия гидроокись
по ГОСТ 4828-83, ч.
Натрий
двууглекислый по ГОСТ 2156-76, ч.
Эфир диэтиловый, х.ч.
Углерод
четыреххлористый по ГОСТ 20288-74.
Спирт этиловый
ректификованный технический по ГОСТ 18300-87.
Вода дистиллированная по ГОСТ 6709-72.
Раствор 1: раствор
лантана азотнокислого, содержащий 0,2 г лантана в 1 см3, 312 г ![]() ,
растворяют при нагревании в 170 см3 концентрированной азотной кислоты, раствор
охлаждают до комнатной температуры и переносят в мерную колбу вместимостью 500
см3, затем доводят до метки дистиллированной водой.
,
растворяют при нагревании в 170 см3 концентрированной азотной кислоты, раствор
охлаждают до комнатной температуры и переносят в мерную колбу вместимостью 500
см3, затем доводят до метки дистиллированной водой.
Раствор 2: смесь
кислот для полирования и растворения образцов кремния. Готовят
в полиэтиленовой банке вместимостью 500 см3 из концентрированных азотной и
фтористоводородной кислот в соотношении 3:1 по объему.
Определяют
содержание фтор-иона в приготовленной смеси кислот: в
четыре стакана вместимостью по 50 см3 приливают по 10 см3 раствора 1 и
нагревают на электроплитке до кипения. В кипящий раствор 1 приливают по 5 см3
раствора 2. Образовавшийся в каждом стакане осадок фторида лантана отделяют
центрифугированием, промывают 5 М азотной кислоты, этиловым спиртом с эфиром,
эфиром и высушивают до постоянной массы. За 100%-ный
выход фторида лантана принимают среднее арифметическое масс четырех выделенных
осадков.
Аммиачный
раствор хлорида бария, содержащий 0,14 г бария в 1 см3 250 г ![]() растворяют в дистиллированной воде, добавляют 91 см3 25%-ного
раствора аммиака, переносят в мерную колбу вместимостью 1000 см3 и доводят до
метки дистиллированной водой. Раствор переносят в стакан вместимостью 2000 см3
и доводят до кипения. Охлажденный раствор фильтруют и хранят в герметически
закрытой посуде. Если при хранении раствора выпадает осадок, раствор необходимо
повторно прокипятить и осадок отфильтровать.
растворяют в дистиллированной воде, добавляют 91 см3 25%-ного
раствора аммиака, переносят в мерную колбу вместимостью 1000 см3 и доводят до
метки дистиллированной водой. Раствор переносят в стакан вместимостью 2000 см3
и доводят до кипения. Охлажденный раствор фильтруют и хранят в герметически
закрытой посуде. Если при хранении раствора выпадает осадок, раствор необходимо
повторно прокипятить и осадок отфильтровать.
Галлий-индиевый сплав, близкий по составу к эвтектике (от 20 до 30% индия и от 80 до
70% галлия): в фарфоровую чашку помещают смесь галлия и индия в отношении 7:3
по массе, нагревают на электрической плитке в течение 2 - 2,5 ч и осаждают на
воздухе.
3.
ПОДГОТОВКА К АНАЛИЗУ
3.1.
Подготовка анализируемых пластин кремния и образцов сравнения к облучению
3.1.1. Для
получения одного результата анализа необходимо подготовить и облучить не менее
двух параллельных пластин кремния. На анализ готовят пластины кремния в виде
квадратов со стороной 12 - 14 мм и толщиной 0,3 - 3,5 мм. Поверхность
отобранных на анализ пластин должна быть химически полированной. Для получения
химически полированной поверхности кремний сначала обрабатывают на абразивных
порошках М28, М20, М14, переходя постепенно от более грубого порошка М28 к М14.
При ручной шлифовке абразивный порошок наносят на плоские стеклянные пластины и
смачивают водой. Для каждого номера порошка должна быть отдельная пластина.
Порошками обрабатывают обе плоскости пластины кремния. Боковую поверхность не
обрабатывают. После шлифовки на каждом порошке на поверхностях не должно быть
царапин, пластины кремния следует тщательно отмывать от предыдущего порошка.
Смесь кислот для
химического полирования (раствор 2) в количестве, достаточном для полного
погружения пластин, наливают в чашку из фторопласта. Пластину кремния зажимают
в пинцет из фторопласта и погружают в полирующую смесь. В процессе полирования
смесь непрерывно перемешивают. Полирование заканчивают, когда поверхность
пластины кремния становится блестящей. Кремний быстро извлекают из полирующей
смеси, промывают проточной водой и подсушивают фильтровальной бумагой. Если на
поверхности обнаруживаются царапины, поры или раковины, обработку следует
повторить.
Образцы, с
поверхности которых перечисленные выше дефекты при повторной обработке не
устраняются, не пригодны для анализа.
Отобранные на
анализ пластины кремния взвешивают, измеряют микрометром толщину в геометрическом
центре, измеряют геометрические размеры пластины штангенциркулем и рассчитывают
площадь поверхности.
Перед установкой в
медную шайбу на плоскость пластины кремния, противоположную облучаемой, наносят
с помощью деревянной палочки индий-галлиевую эвтектику.
Смоченной поверхностью пластину кремния кладут на поверхность массивной медной
шайбы по ее геометрическому центру, слегка прижимают и притирают к ней,
выступившие капельки эвтектики удаляют деревянной палочкой, после чего на
пластину кремния помещают алюминиевую диафрагму и фиксируют положение
прижимными винтами. Отверстие диафрагмы должно быть на 1 - 2 мм меньше диаметра
или стороны квадрата пластины.
3.1.2. Медные шайбы
с образцами сравнения готовят следующим образом: по центру медной шайбы
помещают образец сравнения (кварц, графит или нитрид алюминия) и закрепляют его
диафрагмой и прижимными винтами.
Если необходимо
уменьшить энергию ионов, между диафрагмой и образцом сравнения помещают фольгу
из алюминия, толщина которой должна соответствовать требуемому уменьшению
энергии.
3.2. Облучение
пластин кремния и образцов сравнения
Подготовленные
медные шайбы с пластинами кремния загружают в устройство для облучения,
периодически и в произвольном порядке вводя между ними медные шайбы с образцами
сравнения.
На каждую
определяемую примесь должно быть подготовлено по 9 шайб с образцами сравнения,
облучаемых при трех разных значениях энергии ионов в
интервале от 6 до 10 МэВ для ![]() и от 4 до 6,5 МэВ для протонов.
и от 4 до 6,5 МэВ для протонов.
При определении кислорода
и углерода медные шайбы с пластинами кремния и образцами сравнения (кварц и
графит) облучают ионами ![]() с энергией от 12,7 до 13 МэВ.
с энергией от 12,7 до 13 МэВ.
При определении
азота пластины кремния и образцов сравнения из нитрида алюминия облучают
протонами с энергией 6,5 МэВ.
При облучении
пластин кремния ток однозарядных ионов составляет 1 - 5 мкА, продолжительность
облучения от 20 до 120 мин.
Образцы сравнения
облучают током не более 0,1 мкА, продолжительность облучения 1 - 3 мин.
3.3. Обработка
облученных образцов сравнения
Медные шайбы с
облученными образцами сравнения из кварца выдерживают в расфасовочном боксе в
течение 4 - 5 ч, шайбы с графитом или нитридом алюминия выдерживают 1,5 - 2 ч.
После выдержки
образцы сравнения извлекают из шайб. Пластины кварца протирают ватным тампоном,
смоченным спиртом, упаковывают в алюминиевые кассеты и передают на спектрометр ![]() -совпадений
для измерения кривых распада.
-совпадений
для измерения кривых распада.
3.4.
Обработка пластин кремния после облучения
После окончания
облучения медные шайбы с пластинами кремния поступают в расфасовочный бокс
радиохимической лаборатории, где их выдерживают в течение 5 - 10 мин (при
анализе с радиохимическим выделением аналитических изотопов ![]() или
или ![]() ), или
30 - 45 мин (при инструментальном анализе).
), или
30 - 45 мин (при инструментальном анализе).
По окончании
выдержки пластины кремния извлекают из медных шайб, очищают поверхность ватным
тампоном, смоченным спиртом, и помещают в бокс для химической обработки.
При определении
кислорода и углерода с поверхности облученных пластин кремния должен быть
удален слой толщиной 45 - 50 мкм, которому соответствует уменьшение энергии ионов от 13 до 10 МэВ.
При определении
азота (на протонах) с облученных протонами пластин кремния удаляют слой
толщиной 15 - 20 мкм.
Для удаления
поверхностного слоя пластины кремния обрабатывают в смеси фтористоводородной и
азотной кислот (раствор 2). По 10 - 15 см3 этой смеси наливают в две чашки из
фторопласта. Пластину кремния зажимают в пинцет из фторопласта и погружают в
чашку 1 так, чтобы в процессе травления она была полностью погружена в смесь.
Во время травления смесь постоянно перемешивают.
Пластину кремния
обрабатывают в чашке 1 в течение фиксированного времени (30 или 60 с), затем
быстро извлекают ее из чашки, тщательно промывают водой, подсушивают
фильтровальной бумагой и взвешивают. По разности между массой образца до
травления и после травления определяют толщину стравленного слоя (![]() ), мкм, по формуле
), мкм, по формуле
где ![]() ;
;
![]() - масса образца до травления, мг;
- масса образца до травления, мг;
![]() - масса образца после травления, мг;
- масса образца после травления, мг;
S - полная поверхность
пластины кремния, мм2.
Если после
обработки в чашке 1 толщина стравленного слоя меньше требуемой, травление
продолжают в чашке 2 с учетом скорости травления пластины кремния в чашке 1.
Отработанные растворы сливают в сборники радиоактивных отходов.
По окончании
травления пластину кремния быстро извлекают из смеси кислот, тщательно
промывают водой, подсушивают, взвешивают и определяют полную толщину
стравленного слоя по формуле (1). Толщина стравленного слоя должна быть такой,
чтобы активность оставшейся на пластине части облученного слоя соответствовала
энергии частиц 10 МэВ.
Обработанные таким
образом пластины кремния передают на инструментальный анализ или подвергают
дальнейшей радиохимической обработке.
4.
ПРОВЕДЕНИЕ АНАЛИЗА
4.1.
Инструментальное определение кислорода и углерода в кремнии
Инструментальный
метод применяется для определения кислорода, углерода в поликристаллическом
кремнии и в нелегированном кремнии, и в кремнии, выращенном методом бестигельной зонной плавки, а также для определения
кислорода в кремнии, выращенном по методу Чохральского.
После удаления
поверхностного слоя (см. п. 3.4) пластину кремния упаковывают в кассету из
алюминия и передают на спектрометр ![]() -совпадений
для измерения кривых распада.
-совпадений
для измерения кривых распада.
Перед началом
измерений спектрометр ![]() -совпадений
с помощью источника
-совпадений
с помощью источника ![]() настраивают на режим регистрации аннигиляционных
настраивают на режим регистрации аннигиляционных ![]() -квантов,
после чего определяют естественный фон спектрометра.
-квантов,
после чего определяют естественный фон спектрометра.
Измерения
активности пластины кремния (число совпадений в единицу времени) должны быть
начаты через час после окончания облучения. В течение первого часа измерений,
когда регистрируют активность ![]() ,
интервалы между измерениями активности должны составлять 10 - 15 мин, затем
интервалы между измерениями активности могут быть увеличены до 30 - 60 мин.
Время набора совпадений в каждом таком измерении задают таким, при котором
статистическая погрешность (
,
интервалы между измерениями активности должны составлять 10 - 15 мин, затем
интервалы между измерениями активности могут быть увеличены до 30 - 60 мин.
Время набора совпадений в каждом таком измерении задают таким, при котором
статистическая погрешность (![]() ) не
превышала 0,1. Измерения заканчивают, когда активность образца снижается до
величины естественного фона спектрометра.
) не
превышала 0,1. Измерения заканчивают, когда активность образца снижается до
величины естественного фона спектрометра.
Активность образцов
сравнения измеряют на спектрометре ![]() -совпадений
в течение двух - четырех периодов полураспада соответствующего аналитического
изотопа (для
-совпадений
в течение двух - четырех периодов полураспада соответствующего аналитического
изотопа (для ![]()
![]() = 109,8 мин, для
= 109,8 мин, для ![]()
![]() = 20,38 мин). Для регистрации активности
= 20,38 мин). Для регистрации активности ![]() образцы сравнения из графита и нитрида
алюминия измеряют с интервалом 20 - 40 мин,
образцы сравнения из графита и нитрида
алюминия измеряют с интервалом 20 - 40 мин, ![]() -образцы
сравнения из кварца - с интервалом 60 - 120 мин.
-образцы
сравнения из кварца - с интервалом 60 - 120 мин.
По результатам
измерений активности строят на полулогарифмической бумаге кривые распада
радионуклидов ![]() и
и ![]() в пластинах кремния и образцах сравнения. Эти
кривые строят в координатах
в пластинах кремния и образцах сравнения. Эти
кривые строят в координатах ![]() , где
, где ![]() - скорость счета аналитического изотопа, совп./мин, измеренная в момент времени t.
- скорость счета аналитического изотопа, совп./мин, измеренная в момент времени t.
Кривые распада,
построенные по результатам измерений активности пластин кремния, обычно состоят
из двух компонентов с периодом полураспада 20,38 мин (![]() ) и
109,8 (
) и
109,8 (![]() ).
).
Обработка кривых
распада состоит в их графическом разложении на компоненты и нахождении методом
экстраполяции скорости счета каждого аналитического изотопа ![]() (
(![]() ),
), ![]() (
(![]() ) в
момент окончания облучения.
) в
момент окончания облучения.
Если в ходе
обработки кривых распада обнаружено несоответствие периодов полураспада,
найденных в эксперименте, табличным значениям, анализ данной пластины кремния
необходимо повторить с применением радиохимического выделения аналитических
изотопов.
Кривые распада,
построенные по результатам измерений активности образцов сравнения, являются
однокомпонентными и должны соответствовать периоду полураспада ![]() для графита или
для графита или ![]() для кварца. По этим кривым распада определяют
скорость счета
для кварца. По этим кривым распада определяют
скорость счета ![]() (
(![]() , совп./мин) в кварце и скорость счета
, совп./мин) в кварце и скорость счета ![]() (
(![]() , совп./мин) в графите в момент окончания облучения.
, совп./мин) в графите в момент окончания облучения.
Полученные величины
активностей образцов сравнения для трех значений энергии активирующих частиц
используют для построения калибровочных кривых - зависимостей ![]() ,
, ![]() от энергии активирующих частиц Е, МэВ. Эти калибровочные кривые используют для определения
активности образцов сравнения при промежуточных значениях энергии активирующих
частиц, соответствующих фактической толщине удаленного с пластины кремния
поверхностного слоя.
от энергии активирующих частиц Е, МэВ. Эти калибровочные кривые используют для определения
активности образцов сравнения при промежуточных значениях энергии активирующих
частиц, соответствующих фактической толщине удаленного с пластины кремния
поверхностного слоя.
Массовую долю
примеси в процентах вычисляют по формулам:
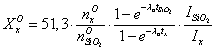 ; (2)
; (2)где ![]() ,
, ![]() - скорость счета радиоизотопа
- скорость счета радиоизотопа ![]() и
и ![]() ,
соответственно в момент окончания облучения в пластине кремния, соответствующая
энергии частиц
,
соответственно в момент окончания облучения в пластине кремния, соответствующая
энергии частиц ![]() , совп./мин;
, совп./мин;
![]() ,
, ![]() - скорость счета радиоизотопа
- скорость счета радиоизотопа ![]() и
и ![]() ,
соответственно в момент окончания облучения в кварце (
,
соответственно в момент окончания облучения в кварце (![]() ) или
графите (С), соответствующая энергии частиц
) или
графите (С), соответствующая энергии частиц ![]() , совп./мин;
, совп./мин;
![]() ,
, ![]() ,
, ![]() - средний ток активирующих частиц при
облучении на ускорителе кремния (x), кварца (
- средний ток активирующих частиц при
облучении на ускорителе кремния (x), кварца (![]() ) или
графита (С), мкА;
) или
графита (С), мкА;
![]() ,
, ![]() ,
, ![]() - продолжительность облучения на ускорителе
кремния (x), кварца (
- продолжительность облучения на ускорителе
кремния (x), кварца (![]() ) и
графита (С), мин.
) и
графита (С), мин.
Описанный принцип
измерения кривых распада, обработки результатов измерения и получения конечного
результата - концентрации определяемой примеси в пластине кремния, - положены в
основу автоматизированной системы "АКАН" в комплексе с ЭВМ ЕС-1010.
Программа обработки результатов измерений составлена на языке
"FORTRAN-4".
За результат
анализа принимают среднее арифметическое двух вычисленных по формуле (2) или
(3) результатов параллельных определений, каждый из которых получен для одной
из двух пластин кремния, облученных при одном режиме работы ускорителя (без
перестройки по энергии).
Разность большего и
меньшего из двух результатов параллельных определений с доверительной
вероятностью Р = 0,95 не должна превышать величин
абсолютных допускаемых расхождений, приведенных в табл. 1.
──────────────────────┬───────────────┬───────────────────────────
Определяемая примесь │Массовая доля │
Абсолютное допускаемое
│ примеси, %
│ расхождение, %
──────────────────────┼───────────────┼───────────────────────────
│ -3
│ -4
Кислород │1 х 10 │6,3 х 10
│ │
│
-4 │ -5
│1 х 10 │6,3 х 10
│ │
│ -5
│ -6
│1 х 10 │6,3 х 10
│ │
│ -6
│ -7
│1 х 10 │6,3 х 10
│ │
│ -7
│ -7
│5 х 10 │3,0 х 10
│ │
│ -3
│ -4
Углерод │1 х 10 │5,5 х 10
│ │
│ -4
│ -5
│1 х 10 │5,5 х 10
│ │
│ -5
│ -6
│1 х 10 │5,5 х 10
│ │
│ -6
│ -6
│2 х 10 │1,0 х 10
Правильность
результатов анализа контролируют методом, заключающимся в проведении анализа
одних и тех же пластин кремния при двух значениях энергий активирующих частиц.
Для контроля правильности отбирают из ранее проанализированных пластин кремния
образцы с содержанием контролируемых примесей на уровне ![]() % по
массе.
% по
массе.
Кислород и углерод
определяют при энергии ионов ![]() 7,5 и 10 МэВ, при определении азота пластины
облучают протонами с энергией 5 и 6,5 МэВ.
7,5 и 10 МэВ, при определении азота пластины
облучают протонами с энергией 5 и 6,5 МэВ.
Результаты анализа
считают правильными с доверительной вероятностью Р =
0,95, если разность между найденными при разных энергиях частиц значениями
концентраций примесей не превышает величин абсолютных допускаемых расхождений,
приведенных в табл. 1.
4.2. Анализ с
применением радиохимического выделения аналитических изотопов
4.2.1.
Определение кислорода в кремнии с применением радиохимического выделения
Метод применяют для
определения содержания кислорода в кремнии, когда необходимо устранить влияние
примесей-источников позитронной активности. Наличие помех от примесей и
необходимость их устранения устанавливают по результатам обработки кривых
распада при анализе кремния инструментальным методом в соответствии с п. 4.1.
Для
определения содержания кислорода с применением радиохимического выделения ![]() пластины кремния после инструментального
анализа по п. 4.1 должны быть вновь обработаны в соответствии с п. 3.1,
повторно облучены, выдержаны после окончания облучения в течение 5 - 10 мин и
обработаны в соответствии с п. 3.4, после чего пластину кремния переносят в
бокс N 2 для радиохимических работ.
пластины кремния после инструментального
анализа по п. 4.1 должны быть вновь обработаны в соответствии с п. 3.1,
повторно облучены, выдержаны после окончания облучения в течение 5 - 10 мин и
обработаны в соответствии с п. 3.4, после чего пластину кремния переносят в
бокс N 2 для радиохимических работ.
В чашку из
фторопласта наливают 5 см3 смеси концентрированных фтористоводородной и азотной
кислот с известным содержанием фтора (раствор 2) и в нее пинцетом из
фторопласта погружают кремний.
За время
растворения в смеси толщина пластины кремния должна уменьшиться не менее, чем на 300 мкм. Количество кремния, переведенного в
раствор, контролируют по уменьшению массы пластины кремния ![]() , мг с
учетом ее полной поверхности S, мм2 (см. п. 3.4).
, мг с
учетом ее полной поверхности S, мм2 (см. п. 3.4).
В реакционную колбу
прибора для дистилляции фтора (черт. 1) помещают 250 - 300 мг измельченного
силикагеля, колбу закрывают воронкой с притертой пробкой и подключают к
водоструйному насосу, после чего в колбу переливают смесь 2 из фторопластовой
чашки, содержащую растворенный кремний, и приливают 10 - 15 см3
концентрированной серной кислоты.
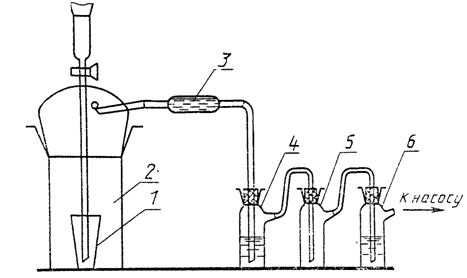
Прибор для
дистилляции соединений фтора с кремнием
из молибденового
стекла

1 - реакционная
колба; 2 - трубка с силикагелем,
пропитанным концентрированной серной кислотой;
3 - приемник фтора,
содержащий 15 см3
дистиллированной
воды
Черт. 1
Фториды кремния
отгоняют в течение 3 - 5 мин при работающем водоструйном насосе и поглощают
дистиллированной водой в приемнике 3. После окончания отгонки приемник 3
отсоединяют от прибора, а затем отключают водоструйный насос.
Раствор из
приемника 3 переливают в стеклянный стакан вместимостью 100 см3, содержащий 15
см3 нагретого до кипения раствора нитрата лантана (раствор 1) и продолжают
нагревание еще в течение 1 мин, непрерывно перемешивая раствор.
Образовавшийся
осадок фторида лантана отделяют центрифугированием, промывают последовательно
горячей 5 М азотной кислотой, спиртом с эфиром, эфиром и затем подсушивают,
помещая пробирку с осадком вблизи электроплитки.
Длительность
химического выделения осадка фторида лантана не превышает 20 - 30 мин.
Высушенный осадок
фторида лантана переносят на кальку, заворачивают в нее, кальку помещают в
алюминиевую кассету и передают на спектрометр ![]() -совпадений
для измерения кривой распада.
-совпадений
для измерения кривой распада.
Активность осадка
фторида лантана измеряют на спектрометре ![]() -совпадений
с интервалами между измерениями 30 - 60 мин. Измерения заканчивают, когда
активность осадка снизится до уровня фона спектрометра.
-совпадений
с интервалами между измерениями 30 - 60 мин. Измерения заканчивают, когда
активность осадка снизится до уровня фона спектрометра.
По результатам
измерений строят кривую распада n = f(t) и по ней определяют скорость счета ![]() во фториде лантана
во фториде лантана ![]() , совп./мин, в момент окончания облучения методом
экстраполяции.
, совп./мин, в момент окончания облучения методом
экстраполяции.
После окончания
измерений осадок фторида лантана извлекают из алюминиевой кассеты и взвешивают.
Химический выход фтора ![]() определяют как отношение массы осадка
определяют как отношение массы осадка ![]() ,
соответствующей 100%-ному выделению фтора, к массе
,
соответствующей 100%-ному выделению фтора, к массе ![]() выделенного в данном опыте осадка
выделенного в данном опыте осадка
![]() . (4)
. (4)
Массовую долю
примеси кислорода (![]() ) в
процентах вычисляют по формуле
) в
процентах вычисляют по формуле
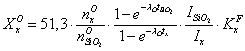 , (5)
, (5)где ![]() - скорость счета радиоизотопа
- скорость счета радиоизотопа ![]() в момент окончания облучения в пластине
кремния, соответствующих энергии частиц
в момент окончания облучения в пластине
кремния, соответствующих энергии частиц ![]() , совп./мин;
, совп./мин;
![]() - скорость счета радиоизотопа
- скорость счета радиоизотопа ![]() в момент окончания облучения в кварце (
в момент окончания облучения в кварце (![]() ), соответствующая
энергии активирующих частиц
), соответствующая
энергии активирующих частиц ![]() , совп./мин;
, совп./мин;
![]() ,
, ![]() - средний ток активируемых частиц при
облучении на ускорителе соответственно кремния (х), кварца (
- средний ток активируемых частиц при
облучении на ускорителе соответственно кремния (х), кварца (![]() ), мкА;
), мкА;
![]() ,
, ![]() - продолжительность облучения на ускорителе
соответственно кремния (х), кварца (
- продолжительность облучения на ускорителе
соответственно кремния (х), кварца (![]() ), мин;
), мин;
![]() - химический выход фтора.
- химический выход фтора.
Измерение кривых
распада выделенных осадков фторида лантана и обработка результатов измерений
предусмотрены программой, составленной для автоматизированной системы
"АКАН" в комплексе с ЭВМ ЕС-1010, написанной на языке
"FORTRAN-4".
За результат
анализа принимают среднее арифметическое двух вычисленных по формуле (5)
результатов параллельных определений, каждый из которых получен для одной из
двух пластин кремния, облученных при одном режиме работы ускорителя (без
перестройки по энергии).
Разность большего и
меньшего двух результатов параллельных определений с доверительной вероятностью
Р = 0,95 не должна превышать величин абсолютных
допускаемых расхождений, приведенных в табл. 2.
Таблица 2
───────────────────────────────┬──────────────────────────────────
Массовая доля примеси │
Абсолютное допускаемое
кислорода, % │ расхождение, %
───────────────────────────────┼──────────────────────────────────
-3 │ -4
1
х 10 │7,0 х 10
│
-4 │ -5
1
х 10 │7,0 х
10
│
-5 │ -6
1
х 10 │7,0 х
10
│
-6 │ -7
1
х 10 │7,0 х
10
│
-7 │ -7
5
х 10 │3,0 х
10
Правильность
результатов анализа контролируют по п. 4.1.
4.2.2. Определение
кислорода и углерода в кремнии с применением радиохимического выделения фтора и
углерода
Метод применяется в
случаях, если необходимо устранить взаимное влияние кислорода и углерода, а
также других примесей - источников позитронной активности на результаты
анализа.
В никелевый тигель
помещают 2 г гидроокиси натрия, 1 г азотнокислого натрия, 0,8 г фторида натрия
и 0,3 г карбоната натрия, тигель ставят в тигельную печь, предварительно
нагретую до 200 - 250 °С, выдерживают в ней до
прекращения газовыделения. Затем тигель извлекают из
печи, охлаждают на воздухе до 100 - 150 °С, после чего
в расплав погружают образец кремния. Температуру в печи повышают до 400 - 450 °С, тигель с образцом ставят в печь и проводят сплавление в
течение 4 - 5 мин. После окончания сплавления никелевый тигель 1 извлекают из
печи, охлаждают на воздухе до 100 - 150 °С и помещают на дно реакционного
сосуда 2 прибора (черт. 2).
1 - никелевый
тигель с охлажденным расплавам;
2 - реакционный
сосуд; 3 - трубка с силикагелем,
пропитанным концентрированной серной кислотой;
4 - приемник фтора,
содержащий 15 см3
дистиллированной
воды; 5 - промежуточная емкость;
6 - приемник
углекислого газа, содержащий 15 см3
аммиачного раствора
хлорида бария
Реакционный сосуд
закрывают капельной воронкой, прибор для дистилляции газов подключают к
водоструйному насосу. В реакционный сосуд через капельную воронку приливают 10
- 15 см3 концентрированной серной кислоты. Через 4 - 5 мин после начала реакции
в колбу приливают 1,5 - 2 см3 воды. Отгонку продолжают еще 3 мин, затем
последовательно отключают приемники 3, 5 и водоструйный насос.
4.2.2.1.
Определение кислорода
Из раствора, содержащегося
в приемнике 4, выделяют осадок фторида лантана по п. 4.2.1.
Химический выход
фтора определяют по формуле
![]() , (6)
, (6)
где ![]() - масса осадка фторида лантана, выделенного в
данном опыте, г.
- масса осадка фторида лантана, выделенного в
данном опыте, г.
Массовую долю
примеси кислорода рассчитывают по формуле (5).
За результат
анализа принимают среднее арифметическое двух вычисленных по формуле (5)
результатов параллельных определений, каждый из которых получен для одной из
двух пластин кремния, облученных при одном режиме работы ускорителя (без
перестройки по энергии).
Разность большего и
меньшего двух результатов параллельных определений с доверительной вероятностью
Р = 0,95 не должна превышать величин абсолютных
допускаемых расхождений, приведенных в табл. 3.
Таблица 3
────────────────────────────────┬─────────────────────────────────
Массовая доля примеси │
Абсолютное допускаемое
кислорода, % │ расхождение, %
────────────────────────────────┼─────────────────────────────────
-3 │ -4
1
х 10 │7,0
х 10
│
-4 │ -5
1
х 10 │7,0
х 10
│
-5 │ -6
1
х 10 │7,0
х 10
│
-6 │ -7
1
х 10 │7,0
х 10
Правильность
результатов анализа контролируют по п. 4.1.
Из раствора,
содержащегося в приемнике 6, выделяют осадок карбоната бария. Для этого
содержимое приемника 6 переливают в стакан. Стакан накрывают часовым стеклом,
ставят на электроплитку и кипятят в течение 1 - 2 мин. Затем раствор охлаждают,
образовавшийся осадок собирают на бумажном фильтре, помещенном на дно воронки Шотта. Фильтрование производят при разряжении, создаваемом
водоструйным насосом. Осадок промывают последовательно холодным раствором
хлорида бария, спиртом, эфиром, подсушивают на воздухе. Фильтр с осадком
переносят на кальку, заворачивают в нее. Кальку с осадком помещают в
алюминиевую кассету для измерения активности на спектрометре ![]() -совпадений";
после экспликации формулы (8) дополнить абзацем: "Измерение кривых распада
выделенных осадков карбоната бария и обработка результатов измерений
предусмотрены программой, составленной для автоматизированной системы
"АКАН" в комплексе с ЭВМ ЕС-1010, написанной на языке
"FORTRAN-4".
-совпадений";
после экспликации формулы (8) дополнить абзацем: "Измерение кривых распада
выделенных осадков карбоната бария и обработка результатов измерений
предусмотрены программой, составленной для автоматизированной системы
"АКАН" в комплексе с ЭВМ ЕС-1010, написанной на языке
"FORTRAN-4".
Активность
выделенного осадка углекислого бария измеряют в течение первого часа через
каждые 10 - 15 мин, затем интервал между измерениями может быть увеличен до 30
- 60 мин. Измерения заканчивают, когда активность углекислого бария уменьшится
до уровня фона в измерительной установке.
По результатам
измерения активности строят кривую распада n = f(t) и по ней методом
экстраполяции определяют скорость счета ![]() в карбонате бария
в карбонате бария ![]() в момент окончания облучения.
в момент окончания облучения.
После окончания измерений
осадок карбоната бария извлекают из алюминиевой кассеты и взвешивают.
Химический выход углерода определяют как отношение массы осадка углекислого
бария, соответствующей 100% выделению введенного в опыт стабильного изотопного
носителя углерода 0,558 г, к массе ![]() выделенного в данном опыте осадка
выделенного в данном опыте осадка
![]() . (7)
. (7)
Массовую долю
примеси углерода (![]() ) в
процентах рассчитывают по формуле
) в
процентах рассчитывают по формуле
где ![]() - скорость счета радиоизотопа
- скорость счета радиоизотопа ![]() в момент окончания облучения в пластине
кремния, соответствующая энергии частиц
в момент окончания облучения в пластине
кремния, соответствующая энергии частиц ![]() , совп./мин;
, совп./мин;
![]() - активность радиоизотопа
- активность радиоизотопа ![]() в момент окончания облучения в графите (С) при
энергии активирующих частиц
в момент окончания облучения в графите (С) при
энергии активирующих частиц ![]() , совп./мин;
, совп./мин;
![]() ,
, ![]() - средний ток активирующих частиц при
облучении соответственно кремния (x) и графита (С), мкА;
- средний ток активирующих частиц при
облучении соответственно кремния (x) и графита (С), мкА;
![]() ,
, ![]() - продолжительность облучения соответственно
кремния (x) и графита (С), мин;
- продолжительность облучения соответственно
кремния (x) и графита (С), мин;
![]() - химический выход углерода.
- химический выход углерода.
За результат
анализа принимают среднее арифметическое двух вычисленных по формуле (8)
результатов параллельных определений, каждый из которых получен для одной из
двух пластин кремния, облученных при одном режиме работы ускорителя (без
перестройки энергии).
Разность большего и
меньшего из двух результатов параллельных определений с доверительной
вероятностью Р = 0,95 не должна превышать величин
абсолютных допускаемых расхождений, приведенных в табл. 4.
Таблица 4
───────────────────────────────┬──────────────────────────────────
Массовая доля примеси │ Абсолютное
допускаемое
углерода, % │ расхождение, %
───────────────────────────────┼──────────────────────────────────
-3
│ -4
1
х 10 │8,3 х
10
│
-4 │ -5
1
х 10 │8,3 х
10
│
-5 │ -6
1
х 10 │8,3 х
10
│
-6 │ -7
2
х 10 │8,3 х
10
Правильность
результатов анализа контролируют по п. 4.1.
4.2.3. Определение
азота в кремнии с применением радиохимического выделения углерода
Метод применяют для
определения содержания азота в кремнии, если массовая доля бора в нем не
превышает ![]() %.
%.
При определении
азота из прибора для дистилляции (черт. 2) исключить приемник 4, соединив
трубку с силикагелем 3 и промежуточную емкость 5 хлорвиниловой трубкой.
Определение выполняют как указано в п. 4.2.2.2.
За результат
анализа принимают среднее арифметическое двух вычисленных по формуле (8)
результатов параллельных определений, каждый из которых получен для одной из
двух пластин кремния, облученных при одном режиме работы ускорителя (без
перестройки по энергии).
Разность большего и
меньшего двух результатов параллельных определений с доверительной вероятностью
Р = 0,95 не должна превышать величин абсолютных
допускаемых расхождений, приведенных в табл. 5.
Таблица 5
───────────────────────────┬──────────────────────────────────────
Массовая доля примеси │Абсолютное допускаемое расхождение, %
азота, % │
───────────────────────────┼──────────────────────────────────────
-3 │ -4
1
х 10 │9,5 х 10
│
-4 │ -5
1
х 10 │9,5 х 10
│
-5 │ -6
1
х 10 │9,5 х 10
Правильность
результатов анализа контролируют по п. 4.1.
ТЕХНОРМАТИВЫ ДЛЯ СТРОИТЕЛЕЙ И ПРОЕКТИРОВЩИКОВ
Copyright © www.docstroika.ru, 2013 -
2026